

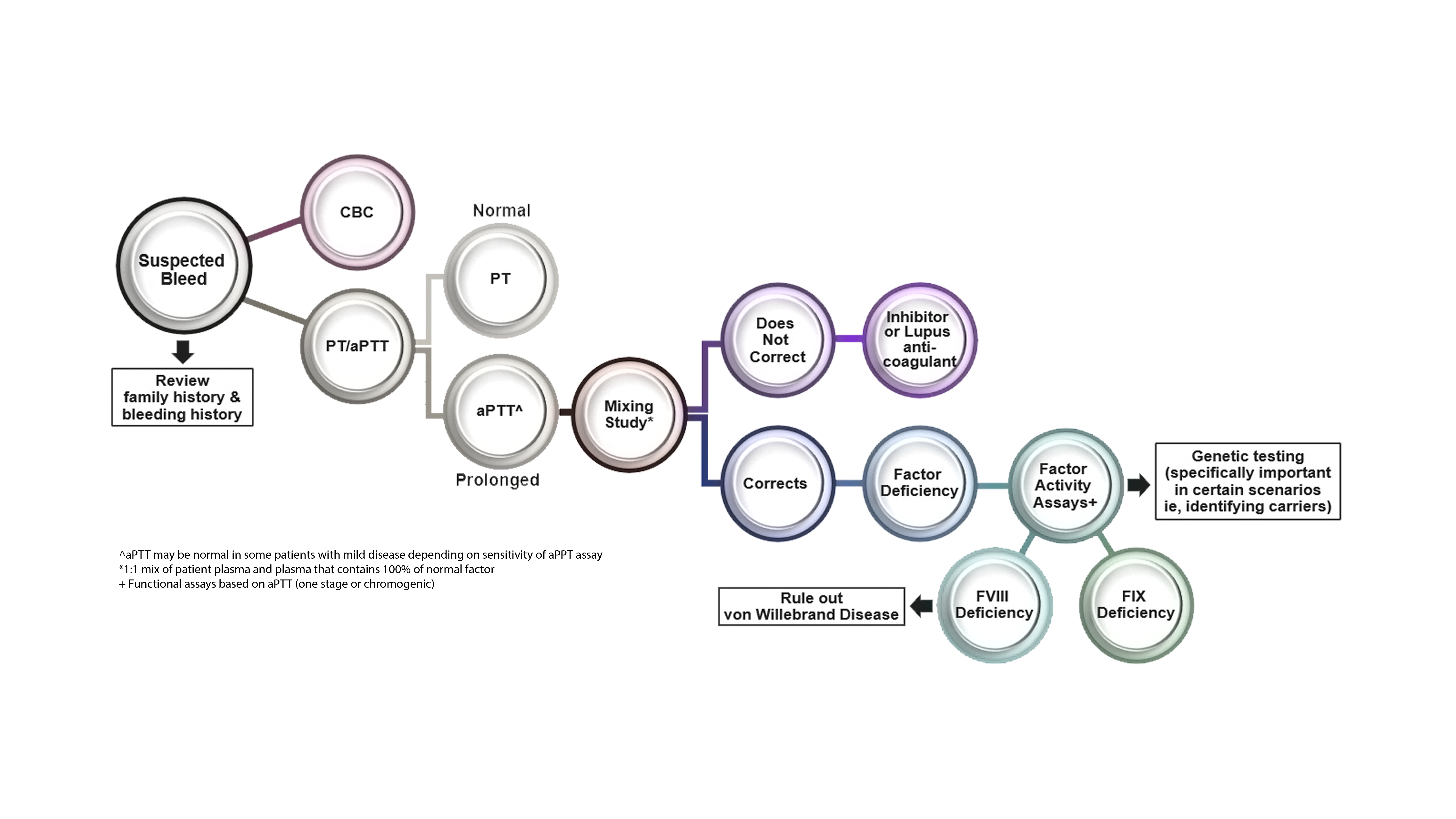
Hemophilia A and B

Hemophilia is a predominately male, X-linked genetic disorder.
Inherited or spontaneous mutations in the F8 (hemophilia A) or F9 (hemophilia B) gene result in a deficiency or absence of functional coagulation factor VIII (FVIII) or coagulation factor IX (FIX), respectively.
70% of hemophilia patients inherit the faulty gene; however, 30% are spontaneous and occur in patients with no family history of the disease.
The majority of carriers (XX) are asymptomatic.
Hemophilia A
Estimated prevalence at birth is 24.6 cases per 100,000 males for all severities of hemophilia A (9.5 cases for severe hemophilia A).
Hemophilia B
Estimated prevalence at birth is 5.0 cases per 100,000 males for all severities of hemophilia B (1.5 cases for severe hemophilia B).
Mild
>5% - 40% of Normal Activity Levels
- Spontaneous bleeding is rare
- Often diagnosed late
- Severe bleeding with major trauma or surgery
Moderate
1% - ≤5% of Normal Factor Activity Levels
- Occasional spontaneous bleeding
- Bleeding into joints and muscles after minor injuries
- Excessive/prolonged bleeding after minor injuries, surgeries, or trauma
Severe
<1% of Normal Factor Activity Levels
- Frequent spontaneous bleeding (~2-5 per month)
- Spontaneous bleeds into joints and soft tissues
- Excessive/prolonged bleeding after minor injuries, surgeries, trauma, or dental work
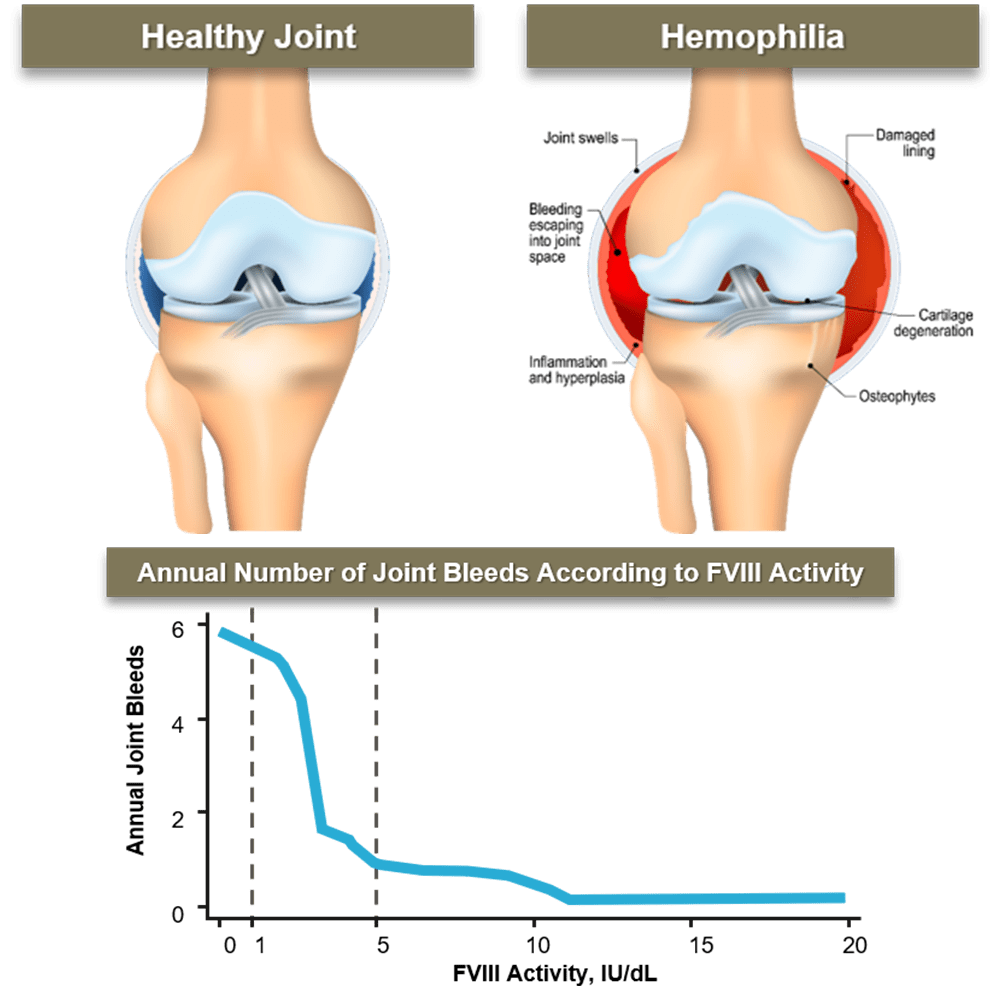
Joint bleeds account for 70-80% of bleeds in patients with hemophilia.
As little as one joint bleed can lead to irreversible joint damage.
Prophylaxis initiated prior to the onset of arthropathy has been shown to preserve joint function in adulthood.
High trough levels are important to prevent bleeds and protect joints; in patients with hemophilia A, those with low factor levels (<5%) have the highest risk for joint bleeds, while those with factor levels of ≥15% had a very low risk approximating no expected joint bleeds.
.
Hemophilia A
FVIII concentrates are the treatment of choice.
Each unit of FVIII/kg infused will raise the plasma FVIII level approximately 2 IU/dL.
Desmopressin (DDAVP) may be utilized in patients with mild or moderate disease.
Individual response should be tested prior to therapeutic use (significant differences between individuals).
Hemophilia B
FIX concentrates are the treatment of choice.
The use of pure FIX concentrates is preferable for treatment over PCC.
Each unit of FIX/kg infused will raise the plasma FIX level approximately 1 IU/dL.
Category Treatment
On Demand Episodic replacement therapy given at the time of clinically evident bleeding
Prophylaxis Continuous/regular replacement therapy given to avoid bleeding
Prevention Replacement therapy given to prevent bleeding for certain high risk periods (eg, surgery)